Cornelia de Lange syndrom





Obsah článku
- Klinické příznaky
- Popis typických příznaků syndromu
- Příčiny vzniku nemoci a dědičnost
- Diagnostika
- Možnosti prenatální diagnostiky
- Léčba
- Prognóza u pacientů se syndromem Cornelia de Lange
Pravděpodobně je nedostatečně diagnostikován, protože u postižených jedinců s mírnými nebo neobvyklými rysy nemusí být syndrom Cornelia de Lange (dále CdLS) nikdy rozpoznán. Výskyt 1:10 000 - 30 000 řadí toto onemocnění mezi takzvaná vzácná onemocnění (anglicky rare diseases).
Klinické příznaky
Klinické příznaky a potíže u pacientů s CdLS mohou být velmi variabilní, od mírných příznaků až po velmi těžké. Typickými projevy u pacientů jsou porucha růstu/neprospívání, kognitivní/mentální deficit, kraniofaciální dysmorfie (nápadné rysy tváře) a nadměrné ochlupení (hirsutismus). Obecně se odlišují dvě formy CdLS: takzvaná mírná, a klasická. Většina typických popisů tohoto syndromu se vztahuje ke klasické formě, ale děti s mírnou formou mohou pociťovat stejné příznaky v menší míře (mild forma-CdLS).
Prvním příznakem, který může být patrný již před narozením, je nedostatečný růst plodu, takzvaná růstová restrikce, což následně vede u novorozence k hypotrofii, kdy váha a výška v době porodu se nachází pod 10. percentilem dle růstových novorozeneckých grafů. Neprospívání a porucha růstu jsou typické i v postnatálním období.
Popis typických příznaků syndromu
1. kraniofaciální dysmorfie: příznaky v oblasti obličeje a hlavy
- menší obvod hlavy (mikrocefalie)
- srostlé a husté obočí (synofrys), klenuté obočí
- u dětí jsou nápadné dlouhé tlusté řasy
- ptóza víčka (pokles)
- oči jsou uloženy dále od sebe takzvaný (hypertelorismus)
- z dalších očních příznaků: malá rohovka (mikrokornea), malformace slzných kanálů, strabismus, nystagmus, blefaritis, krátkozrakost
- vpáčený kořen nosu, anteverze nostril
- dlouhé filtrum, úzký horní ret, svěšené koutky ústní
- vysoké klenuté patro, rozštěpy, typická je pozdní erupce zubů a anomálie zubů
- tlusté dysplastické dozadu rotované ušní boltce
- menší brada (mikrognatie).
2. kožní projevy:
- nadměrné ochlupení (hirsutismus)
- hypoplastické bradavky a pupečník
- jedna příčná dlaňová rýha (palmární)
3. končetinové vady:
- malé končetiny (mikromelie)
- fokomélie – jedná se o defekt končetiny, kdy dochází k poruše vývoje kostí končetin, dochází k nasedání ruky či nohy přímo na pletenec ramenní či pánevní.
- menší počet prstů (oligodaktylie)
- rohlíčkovité zahnutí malíčků ( klinodaktylie)
- srůst druhého a třetího prstu (syndaktylie)
- z dalších: proximálně postavený palec, flekční kontraktury v kloubech a další.
4. z dalších vrozených vývojových vad:
- srdeční vady – až u třetiny pacientů
- anomálie ledvin, hypoplázie genitálu, u chlapců nesestouplá varlátka (kryptorchismus)
- idiopatická trombocytopenická purpura
Hlavním příznakem je různý stupeň opoždění psychomotorického vývoje. IQ se uvádí mezi 30–86, průměrně kolem 53. S tímto souvisí i opožděný vývoj řeči, poruchy chování ve smyslu autistických rysů, prvky hyperaktivity, a u některých se mohou objevit i křeče (epilepsie).
Příčiny vzniku nemoci a dědičnost
Nejčastěji se narodí dítě s CdLS zdravým rodičům, následkem takzvaně de novo vzniklé mutace. Nemoc CdLS vzniká obvykle jako výsledek náhodné genetické změny (mutace) v jednom z několika důležitých genů: nejčastěji v genu NIPBL. Mutace v tomto genu identifikujeme u více než poloviny všech lidí s onemocněním CdLS. Mutace v jiných genech, jejichž příčinou je rovněž CdLS, se v menší míře týká i genů: SMC1A, HDAC8, RAD21, SMC3 a dalších, kde jsou nálezy raritní.
Asi v patnácti procentech případů zůstane příčina vzniku syndromu Cornelia de Lange u pacienta neznámá. Výzkumné týmy stále hledají další změny ve známých genech stejně jako varianty v jiných genech, které mohou způsobit tuto nemoc.
Pokud syndrom CdLS způsobují varianty genu NIPBL, RAD21 nebo SMC3, jedná se o dědičnost autozomálně dominantní (AD). AD dědičnost znamená, že každý zdravý organizmus potřebuje ke správné funkci obě alely genu a jedna kopie změněného genu (mutace) v každé buňce už způsobí vznik nemoci. Naštěstí, většina případů CdLS vzniká jako důsledek nových genových variant (de novo). Pro potomky pacienta (muže nebo ženy) s CdLS je 50% riziko přenosu do další generace bez ohledu na pohlaví (buď dojde prostřednictvím vajíčka nebo spermie k předání zdravé alely genu/ „zdravého genu“, nebo naopak předání genu s mutací/„nemocného genu“).

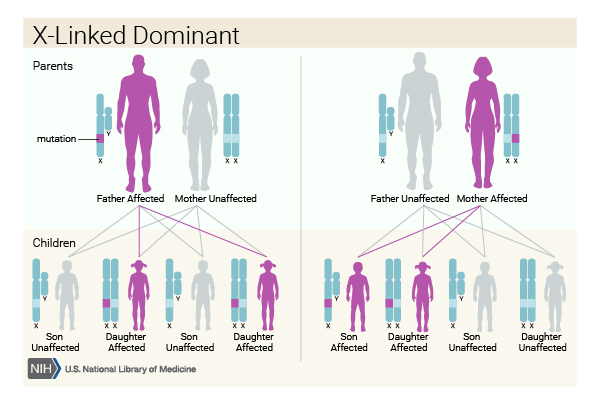
V případě, že je syndrom CdLS způsoben variantami v genu HDAC8 nebo SMC1A , jedná se o dominantní dědičnost vázanou na chromozom X, takzvaně X-vázaná dominantní dědičnost XD. XD dědičnost je odlišná u mužů a u žen (muži mají jenom jeden X chromozom, zatímco organizmus ženy obsahuje dva X chromozomy). U muže tedy stačí jedna kopie změněného genu (mutace) na jeho jediném X chromozomu a postižený muž již onemocní syndromem CdLS. Žena, která má přítomnou mutaci na jednom ze svých dvou X chromozomů, bude spíše přenašečkou onemocnění, vzácně může mít velmi mírné příznaky onemocnění CdLS ve srovnání s postiženým mužem, který má závažné příznaky nemoci. Většina případů je ale opět vázaných na X chromozom, je výsledkem de novo variant v genu HDAC8 nebo SMC1A a nevyskytuje se v rodině (narodí dvěma zdravým rodičům).
Pro potomky pacientky/ženy s CdLS s mutací v genech HDAC8 nebo SMC1A platí v případě XD dědičnosti odlišná rizika pro dcery a syny. Pokud předá svým synům zdravou alelu genu/“zdravý chromozom X“, 50 % jejích synů bude zdravých. Předáním chybné alely/„poškozeného chromozomu X“, zdědí 50 % synů závažnou formu nemoci CdLS. Pro její dcery platí, že pokud předá alelu genu na chybný/ poškozený chromozom Xm,“ 50 % dcer zdědí mírnou formu CdLS nebo budou přenašečky. Předáním „zdravého“ genu na „zdravý“ X chromozom bude 50 % dcer úplně zdravých a vlohu na X chromozomu nepřenesou ani do další generace.
Pro potomky pacienta/muže s CdLS s mutací v genech HDAC8 nebo SMC1A platí v případě XD dědičnosti pro jeho dcery a syny také odlišná rizika: muž s CdLS předává obecně, jako každý jiný zdravý muž, všem svým synům vždy chromozom Y a nepředává jim gen X, proto všichni jeho synové budou zdrávi. Dcerám předává pokaždé svůj jediný chromozom X, na kterém je přítomná mutace, proto všechny jeho dcery zdědí jen mírnou formu CdLS nebo budou jen přenašečkami nemoci.
Rodiče, kterým se narodí dítě s de novo mutací v kterémkoliv genu pro CdLS mají pro svoje další dítě riziko mírně zvýšené. Je to z důvodu, že některá vajíčka nebo některé spermie (to nelze vyšetřit) nesou takzvanou zárodečnou (germinální) mutaci. Ostatní spermie nebo vajíčka ji nenesou a jsou zdravé. Tomuto jevu odborně říkáme zárodečná mozaika, a je popisována v 3,5–5,4 %. Riziko postižení pro dalšího potomka je zvýšené v důsledku možnosti existence této takzvané zárodečné mozaiky u některého z rodičů.
Diagnostika
V minulosti se diagnostika opírala jen o přítomnost klinických příznaků specifických pro tento syndrom. S rozvojem molekulárně genetického testování se nám nyní daří diagnostikovat i pacienty, u kterých jsme tento syndrom předpokládali, ale i u těch, kteří mají například jen mírné příznaky nebo příznaky, které se mohou navzájem překrývat s dalšími genetickými syndromy.
Pro klinickou diagnostiku CdLS syndromu byla vytvořena škála sčítající body za klasické (hlavní) fenotypové projevy a vedlejší fenotypové projevy spojené s CdLS. Skóre >11 značí klasický fenotyp CdLS, pokud jsou přítomny alespoň tři hlavní rysy, 9-10 značí neklasický fenotyp, pokud jsou přítomny alespoň dva hlavní rysy, >4 stačí k molekulárnímu otestování CdLS, pokud je přítomen alespoň jeden hlavní znak. Při podezření na CdLS se genetické vyšetření provádí z odběru krevního vzorku od pacienta a následuje podrobná analýza jeho DNA. U dětí do 18 let věku musí s vyšetřením po genetické konzultaci a vysvětlení souhlasit jeho rodiče.
V současné době k určení definitivní diagnózy používáme takzvané sekvenování nové generace (zkratka NGS – Next Generation Sequencing). Pro představu, se jedná o vyšetření, kdy jsme schopni u pacienta v rámci jednoho vyšetření vyšetřit naráz několik stovek a tisíců genů najednou, v minulosti se vyšetřovaly pouze jednotlivé geny.
S rozvojem analýz bylo postupně s tímto syndromem popsáno několik genů, které jsou zodpovědné za rozvoj onemocnění, z nichž nejčastěji nacházíme patogenní varianty v genu NIPBL (60 %), z dalších méně častých genů jsou to geny SMC1A, HDAC8, RAD21, SMC3 (5–10 %). Pomocí tohoto molekulárně genetického vyšetření, které plně hradí zdravotní pojišťovny, jsme schopni stanovit diagnózu CdLS u cca 85 % pacientů. I přes nejnovější metody analýz zůstává část pacientů (asi 15 %) bez nalezené příčiny na genetické úrovni, i když splňuje kritéria pro tento syndrom. Může to být způsobeno tím, že dosavadní metody nezachytí mutaci, mutace se může nacházet v oblasti genu, která unikne analýze a/nebo mohou být samozřejmě ve hře doposud neobjevené geny. U některých pacientů byla příčina CdLS spojena s mutacemi v genech BRD4, MAU2 a ANKRD11.
Následkem mutace neumí organizmus produkovat správně fungující proteiny a vznikají defektní proteiny, nebo jejich nedostatečné množství. Proteiny produkované většinou genů zapojených do syndromu Cornelia de Lange přispívají ke struktuře nebo funkci kohezinového komplexu, což je skupina proteinů s důležitou rolí v řízení vývoje před narozením. Tento komplex pomáhá regulovat strukturu a organizaci chromozomů, stabilizovat genetickou informaci buněk a opravovat poškozenou DNA. Reguluje i aktivitu konkrétních dalších genů, které řídí vývoj obličeje, končetin a dalších částí těla. Z toho důvodu mutace ve výše uvedených genech NIPBL, SMC1A, HDAC8, RAD21 a SMC3 způsobují různě závažné příznaky syndromu Cornelia de Lange.
Ze stejné příčiny je spektrum příznaků u jednotlivých pacientů s CdLS s různými mutacemi odlišně závažné, odborně se to označuje termínem genotypovo-fenotypová korelace. Vědci se dále domnívají, že pro určení specifických příznaků a symptomů u každého jedince mohou být důležité i další genetické nebo environmentální faktory (odborně se to nazývá variabilní penetrance a neúplná expresivita, zjednodušeně řečeno, tvorba proteinu z chybného genu a fungování organizmu je ovlivněno i dalšími faktory). Obecně varianty genu SMC1A, RAD21 a SMC3 způsobují mírnější příznaky a symptomy než varianty genu NIPBL. Varianty v genu HDAC8 způsobují poněkud odlišný soubor rysů, včetně opožděného uzavření „měkkého místa“ na hlavě (přední fontanely) v kojeneckém věku, široce postavených očí a zubních abnormalit. Stejně jako postižení jedinci s variantami genu NIPBL mohou mít i ti s variantami genu HDAC8 významné intelektuální postižení.
Možnosti prenatální diagnostiky
Na syndrom nás může upozornit přítomnost nějaké vrozené vývojové vady, či například růstová restrikce plodu, mikrocefalie a další. Diagnostika v prenatálním období je ale vždy složitá, protože výše uvedené vady a symptomy mohou být součástí dalších stovek syndromů a mohou se vyskytovat i izolovaně. Izolované vady jsou navíc nejčastěji multifaktoriální, tedy bez známé genetické příčiny.

Menší vady taktéž nemusí být ultrazvukovými metodami odhalitelné a lze je zpozorovat až po narození. Některé z vad – a samozřejmě opožděný vývoj – jsou rozpoznatelné až v průběhu dětství. Většina dětí s CdLS se také rodí v důsledku de-novo mutace zdravým rodičům, a proto není prenatálně odhalitelná.
Jednoznačně vyšetřitelná diagnóza je u potomků pacientů s CdLS, u kterých je známá konkrétní mutace a je známý typ dědičnosti (viz výše v textu; je velký rozdíl, zda se jedná o AD nebo XD dědičnost) a důležitou roli v případě XD dědičnosti hraje i pohlaví očekávaného dítěte. V tomto případě, opět se souhlasem, lze provést DNA analýzu plodu – z vyšetření choriových klků (takzvaného CVS) nebo plodové vody z amniocentézy (AMC). Rodič s CdLS syndromem pak obdrží přesně určenou míru rizika postižení dítěte.
Rodičům se znalostí genetické mutace lze doporučit i metody asistované reprodukce (IVF – in vitro fertilizace) a preimplantační genetické diagnostiky (PGD – preimplantation genetic diagnosis, též prevent genetic disease), kdy se embrya po úspěšném oplození vyšetří a do dělohy příští maminky se zavede zdravé, které onemocnění nezdědilo. V ČR se zabývá touto problematikou například institut Repromeda či jiná IVF centra.
Léčba
V současné době neexistuje žádná léčba, kterou by bylo možné onemocnění zcela vyléčit. Přístup k pacientovi je velmi individuální, multidisciplinární a odvíjí se od potřeb každého jedince.
Ortopedie nabízí chirurgickou korekci vrozených vývojových vad končetin nebo skoliózy; neurologové sledují psychomotorický vývoj a eventuelně řeší neurologické komplikace jako křeče, poruchy spánku a další. V přítomnosti srdeční vady jsou děti dlouhodobě sledovány v kardiologické ambulanci a pokud to vada vyžaduje, je následně řešena v kardiochirurgických centrech. Vzhledem k častému neprospívání je důležitou součástí týmu gastroenterolog, který řeší problém s výživou a příjmem potravy. V pozdějším dětském věku je důležitá péče psychiatrů a psychologů. Část dětí může trpět poruchami chování, autistickými rysy, hyperaktivitou a podobně. V případě očních komplikací je nutné dlouhodobé sledování očními lékaři. Důležitou roli, a to nejen ve stimulaci vývoje, hraje rehabilitační péče, tedy fyzioterapie, a ergoterapie, a vhodný výběr školy a profese. V dětském věku je velmi důležitá logopedická péče ke zlepšení komunikačních dovedností, ať již verbálních či neverbálních. Jednu z nejvýznamnějších roli v péči a stimulaci dítěte samozřejmě sehrává rodina a nejbližší okolí.
Prognóza u pacientů se syndromem Cornelia de Lange
I když neexistuje cílená léčba ani genová terapie, většina dětí, u kterých je diagnostikován syndrom Cornelia de Lange, se dožívá normální, běžné délky života. Protože většina pacientů má určitou míru mentálního postižení, je běžné, že dítě bude celoživotně odkázáno na péči a dohled (i po dosažení plnoletosti). Při pečlivé dlouhodobé prevenci a léčbě symptomů (příznaků nemoci) vedou však pacienti s CdLS dlouhý, šťastný a normální život.
Podpůrné organizace v České republice
- www.ranapece.cz
- www.centrumprovazeni.cz
- www.vzacnaonemocneni.cz
- https://www.orpha.net/cs/disease/detail/199
Mezinárodní organizace a sdružení pro Cornelia de Lange syndrom
- https://www.cdlsusa.org/
- https://www.cdlsworld.org/xwiki/bin/view/Main/WebHome
- https://www.cdls.org.uk/
- https://www.cdls.org.au/
- https://rarediseases.org/rare-diseases/cornelia-de-lange-syndrome/
- https://www.orphananesthesia.eu/en/rare-diseases/published-guidelines/cornelia-de-lange-syndrome/285-cornelia-de-lange-syndrome/file.html
Související literaturu a další zdroje informací naleznete také v naší Odborné knihovně.
Zaujal Vás článek a chcete každý měsíc dostávat informace o nových příspěvcích? Přihlaste se k odběru newsletteru nebo nás sledujte na Facebooku!



![Hromada starých fotografií [fotograf Rodolfo Clix]](/sites/default/files/styles/article/public/2021-01/xN550,P20-,P20Predci.png,qh=df66a196,aitok=du4n_GB4.pagespeed.ic.IcG3w_0pu2.jpg "Hromada starých fotografií [fotograf Rodolfo Clix]")

![Dívka s bolestmi zad [forograf - Karolina Grabowska]](/sites/default/files/styles/article/public/2020-12/xID,P20161,P20-,P20Revma.png,qitok=zrH0WteF.pagespeed.ic.P-dJnZtkRB.jpg "Dívka s bolestmi zad [forograf - Karolina Grabowska]")








{kind=link}